|
|
|
|
|
3D Genome Browser:实现可视化基因组3D结构和长距离染色体作用 | Genome Biology |
|
|
论文标题:The 3D Genome Browser: a web-based browser for visualizing 3D genome organization and long-range chromatin interactions
期刊: Genome Biology
作者: Yanli Wang, Fan Song, Bo Zhang, Lijun Zhang, Jie Xu, Da Kuang, Daofeng Li, Mayank N. K. Choudhary, Yun Li, Ming Hu, Ross Hardison, Ting Wang and Feng Yue
发表时间:2018/10/4
数字识别码: 10.1186/s13059-018-1519-9
原文链接:https://genomebiology.biomedcentral.com/articles/10.1186/s13059-018-1519-9?utm_source=WeChat&utm_medium=Website_linksSocial_media_organic&utm_content=CelZha-MixedBrand-multijournal-Multidisciplinary-China&utm_campaign=ORG_AWA_CZH_BMCWechat_dailyposts_blogs
微信链接:https://mp.weixin.qq.com/s/oTjxIH9cPhrgrCBlOOLRaw
在哺乳动物的基因组中,染色体的三维结构在基因表达调控中扮演着重要的角色。在DNA层面上,远端的调控原件(如增强子)可以通过空间结构的接近来调控目标基因的表达。在更高一级的染色体结构中,近年来发现的拓扑结构域(topologically associating domains)被认为是哺乳动物染色体结构的基本单元[1]。拓扑结构域是大小为兆碱基的染色体高级结构之一,最早被Hi-C技术所揭示。拓扑结构域为解释远端增强子的基因调控提供了非常好的解释。除了拓扑结构域,其他染色体结构例如A/B结构[2]、染色体环(chromatin loop)[3]等也相应被Hi-C衍生技术所揭示。这些Hi-C衍生技术,例如Hi-C,ChIA-PET[4],Capture Hi-C[5],PLAC-seq[6]和HiChIP[7],为研究染色体空间结构提供了前所未有的机会和挑战。然而,随着Hi-C数据的飞速增长,高效便利的数据可视化技术变得迫在眉睫。同时,高效的可视化对于揭示隐藏在Hi-C数据背后的生物学意义具有重要作用。然而,由于这些数据通常数量庞大且复杂,独立研究者如果想自己实现数据的可视化将变得低效且耗时。
最近来自美国宾州州立大学的岳峰课题组和华盛顿大学的王艇课题组在Genome Biology发表了一篇名为“The 3D Genome Browser: a web-based browser for visualizing 3D genome organization and long-range chromatin interactions”的方法文章。该文章介绍了他们开发的基于网页的3D基因组浏览器(www.3dgenome.org)。这是目前最流行的3D基因组浏览器,迄今为止, 已被来自120多个国家的数万计用户访问过,网页点击量已经超过了数十万次。该网站拥有众多染色体结构相关数据类型,包括Hi-C,GAM,SPRITE,DNase Hi-C,ChIA-PET,PLAC-seq,HiChIP和Capture Hi-C。数据对象包括人类和小鼠的数十种组织和细胞系,总共数据量达到300余个。网站数据加载迅速,可以在5秒之内打开一个10Mb大小区域的Hi-C热图。
该浏览器能够使研究人员更加方便快捷地可视化来自于高通量染色体结构捕获技术(Hi-C)的数据。其主要功能如下:1)使用热图的方式可视化Hi-C数据以及Hi-C类型技术如GAM、SPRITE、和DNase Hi-C;2)可视化不同染色体之间的3D结构;3)比较不同组织或者物种之间的染色体3D构型;4)将Hi-C 数据转换为虚拟4C(virtual 4C),从而可以更方便地查看特定位点和染色体上其他位置的相互作用;5)可视化基于染色质免疫共沉淀结合或染色体区域捕获的Hi-C衍生技术(例如ChIA-PET,PLAC-seq, HiChIP,Capture Hi-C)。
在这个项目里,研究员们提出了一种新的二进制格式(BUTLR)用来保存Hi-C数据,从而极大的减少使文件大小并且提高查询速度。除此之外,该Browser还提供了方便的染色体区域缩放功能。用户可以通过基因,染色体位置,和SNP编号对Hi-C数据进行查询。更为重要的是,用户可以将自己的UCSC或者WashU track和Hi-C热图进行无缝衔接,从而极大地扩展了可以显示地数据类型。
下面我们将用几个例子来说明如何使用3D Genome Browser进行Hi-C数据挖掘。

图1: 3D 基因组浏览器的Hi-C查询页面。用户可以选择网站内建数据,或者上传自己制作的BUTLR文件。
1. 利用Hi-C数据研究染色体相互作用
首先,打开Hi-C查询页面(图1),在该页面中我们可以选择Hi-C方法、物种、参照基因组、组织或细胞系以及数据分辨率。例如,我们可以查询SHH基因在GM12878细胞系中的Hi-C热图。从Hi-C热图上(图2)我们可以看出,该基因与其上游一个已知的增强子区域位于同一TAD内,并且该增强子和SHH启动子存在较高的相互作用,从而证实了该增强子对SHH的调控作用。我们还可以点击页面右上方的柱状图查看该基因在ENCODE中100多个组织中的表达情况。

图2: SHH基因在GM12878细胞系中的Hi-C热图。虚线显示SHH与上游一个增强子区域存在高相互作用,并且该增强子和SHH在同一个TAD中。
2. 利用Virtual 4C、DHS linkage和ChIA-PET研究SNP的目标基因
Hi-C还可以揭示SNP的潜在目标基因。我们利用virtual 4C查询rs12740374,该SNP与人群中低密度脂蛋白升高有关。虚拟4C显示该SNP与其下游SORT1的启动子存在较高作用,这一发现同时也被DNA高敏感性位点连锁(DHS linkage)和ChIA-PET数据所支持(图3)。同时图中下方的组蛋白数据也显示该SNP位于一个可能的增强子区域。这些证据表明SORT1可能是rs12740374的目标基因。

图3: 利用虚拟4C和DHS连锁揭示SNP的潜在目标基因。rs12740374位于一个可能的增强子区域和CEBPB蛋白结合位点。
3. 利用Capture Hi-C数据研究高分辨率启动子-增强子的相互作用
在图4中,我们利用Capture Hi-C数据分析PAX5的调控机制。我们首先发现在原始B细胞中PAX5的启动子和其上游的ZCCHC7区域存在相互作用[13]。通过DNA酶超敏感位点(DHS)数据和组蛋白修饰数据,我们也发现了与该启动子作用的区域是一个可能的增强子。同样的,在Hi-C热图上,我们也发现了这两个区域存在较强的相互作用。这些证据都说明了位于ZCCHC7上游的这个增强子可能是调控PAX5基因表达的关键。有趣的是,之前的研究报道指出该增强子的删除使得PAX5表达下调并导致白血病[14]。这个例子反映了我们可以利用3D Genome Browser和使用Capture Hi-C数据研究精细的启动子-增强子作用。
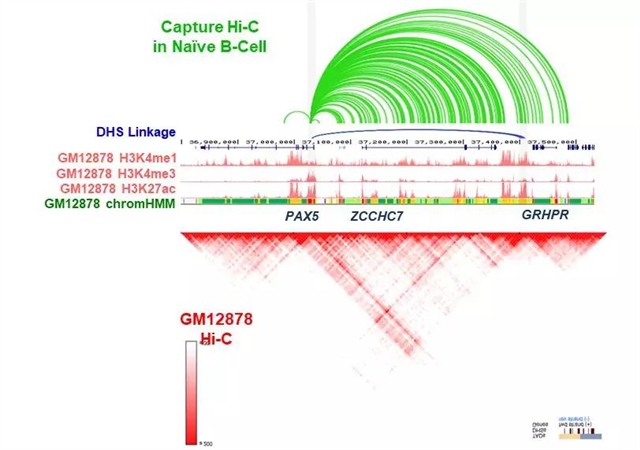
图4: 利用Capture Hi-C数据揭示PAX5的可能调控机制。
4. 通过对比Hi-C热图来研究不同物种间染色质构型的保守程度
我们还可以用Hi-C对比模式来研究染色质构型在不同物种之间的保守性。图5显示了BCL6区域在人淋巴细胞系(GM12878)和鼠细胞系(CH12)的Hi-C热图。从Hi-C热图的相似性可以看出该区域的染色质构型在人和鼠中保守程度较高。

图5: 比较人和小鼠的BCL6基因附近的染色质构型的保守性。
5. 利用Hi-C数据检测染色体结构变异
许多研究发现Hi-C可以被用于检测染色体结构变异(structural variantion)。不同类型的结构变异,例如删失、插入、异位和倒装都会导致Hi-C热图发生相应的变化[15]。BCR-ABL基因融合是慢性粒系白血病的致病变异。该变异是由9号染色体和22号染色体的异位变异导致的。我们可以通过Hi-C热图检测该异位变异(图6)。K562是从慢性粒系白血病患者中培育的细胞系。图6a显示的是K562中22号染色体9号染色体的Hi-C热图。中间菱形区域显示出22号染色体9号染色体存在较强的相互左右,而在正常细胞中,这种染色体之间的相互作用是微乎其微的,并且远小于染色体内部的相互作用。因此,我们能够推断出22号染色体9号染色体发生了融合,导致了不同染色体之间的相互作用增强。并且在融合断点区域,我们可以看到是BCR和ABL基因发生了融合。相比之下,在正常的GM12878淋巴细胞系中,我们没有看到类似的不同染色体之间的相互作用增强(图6b)。通过3D Genome Browser的查询染色体之间的作用功能,研究者能够发现基因组的结构变异及其可能导致的基因调控改变。
3D Genome Browser还有众多功能未能在本文涵盖,例如虚拟4C(virtual 4C)和不同Hi-C数据的差异分析,这些功能同样有助于挖掘Hi-C数据背后的生物学意义。欢迎广大有志于学习3D基因组的科研工作者使用和探索我们的3D Genome Browser。

图6: a).K562中chr22和chr9的Hi-C热图,虚线显示断点在两条染色体所在位置。b).GM12878中正常的chr22和chr9的Hi-C热图没有显示出增强的染色体之间的相互作用。
参考文献:
【1】 Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, Ren B: Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 2012, 485:376-380.
【2】 Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, et al: Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 2009, 326:289-293.
【3】 Rao SS, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, Aiden EL: A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 2014, 159:1665-1680.
【4】 Li G, Fullwood MJ, Xu H, Mulawadi FH, Velkov S, Vega V, Ariyaratne PN, Mohamed YB, Ooi HS, Tennakoon C, et al: ChIA-PET tool for comprehensive chromatin interaction analysis with paired-end tag sequencing. Genome Biol 2010, 11:R22.
【5】 Mifsud B, Tavares-Cadete F, Young AN, Sugar R, Schoenfelder S, Ferreira L, Wingett SW, Andrews S, Grey W, Ewels PA, et al: Mapping long-range promoter contacts in human cells with high-resolution capture Hi-C. Nat Genet 2015, 47:598-606.
【6】 Fang R, Yu M, Li G, Chee S, Liu T, Schmitt AD, Ren B: Mapping of long-range chromatin interactions by proximity ligation-assisted ChIP-seq. Cell Res 2016, 26:1345-1348.
【7】 Mumbach MR, Rubin AJ, Flynn RA, Dai C, Khavari PA, Greenleaf WJ, Chang HY: HiChIP: efficient and sensitive analysis of protein-directed genome architecture. Nat Methods 2016, 13:919-922.
【8】 Zhou X, Maricque B, Xie M, Li D, Sundaram V, Martin EA, Koebbe BC, Nielsen C, Hirst M, Farnham P, et al: The Human Epigenome Browser at Washington University. Nat Methods 2011, 8:989-990.
【9】 Durand NC, Robinson JT, Shamim MS, Machol I, Mesirov JP, Lander ES, Aiden EL: Juicebox Provides a Visualization System for Hi-C Contact Maps with Unlimited Zoom. Cell Syst 2016, 3:99-101.
【10】 Kerpedjiev P, Abdennur N, Lekschas F, McCallum C, Dinkla K, Strobelt H, Luber JM, Ouellette SB, Ahzir A, Kumar N, et al: HiGlass: Web-based Visual Comparison And Exploration Of Genome Interaction Maps. bioRxiv 2017.
【11】 Tang B, Li F, Li J, Zhao W, Zhang Z: Delta: a new web-based 3D genome visualization and analysis platform. Bioinformatics 2018, 34:1409-1410.
【12】 Huang J, Liu X, Li D, Shao Z, Cao H, Zhang Y, Trompouki E, Bowman TV, Zon LI, Yuan GC, et al: Dynamic Control of Enhancer Repertoires Drives Lineage and Stage-Specific Transcription during Hematopoiesis. Dev Cell 2016, 36:9-23.
【13】 Javierre BM, Burren OS, Wilder SP, Kreuzhuber R, Hill SM, Sewitz S, Cairns J, Wingett SW, Varnai C, Thiecke MJ, et al: Lineage-Specific Genome Architecture Links Enhancers and Non-coding Disease Variants to Target Gene Promoters. Cell 2016, 167:1369-1384 e1319.
【14】 Puente XS, Bea S, Valdes-Mas R, Villamor N, Gutierrez-Abril J, Martin-Subero JI, Munar M, Rubio-Perez C, Jares P, Aymerich M, et al: Non-coding recurrent mutations in chronic lymphocytic leukaemia.Nature 2015, 526:519-524.
【15】 Dixon J, Xu J, Dileep V, Zhan Y, Song F, Le VT, Yardimci GG, Chakraborty A, Bann DV, Wang Y, et al: An Integrative Framework For Detecting Structural Variations In Cancer Genomes. Nature Genetics2018.
摘要:
Abstract
Here, we introduce the 3D Genome Browser, http://3dgenome.org, which allows users to conveniently explore both their own and over 300 publicly available chromatin interaction data of different types. We design a new binary data format for Hi-C data that reduces the file size by at least a magnitude and allows users to visualize chromatin interactions over millions of base pairs within seconds. Our browser provides multiple methods linking distal cis-regulatory elements with their potential target genes. Users can seamlessly integrate thousands of other omics data to gain a comprehensive view of both regulatory landscape and 3D genome structure.
阅读论文全文请访问:https://genomebiology.biomedcentral.com/articles/10.1186/s13059-018-1519-9?utm_source=WeChat&utm_medium=Website_linksSocial_media_organic&utm_content=CelZha-MixedBrand-multijournal-Multidisciplinary-China&utm_campaign=ORG_AWA_CZH_BMCWechat_dailyposts_blogs
期刊介绍:
Genome Biology(https://genomebiology.biomedcentral.com/) publishes outstanding research in all areas of biology and biomedicine studied from a genomic and post-genomic perspective.
The current impact factor is 13.214* and the journal is ranked 4th among research journals in the Genetics and Heredity category by Thomson Reuters. Genome Biology is the highest ranked open access journal in the category.
2017 Journal Metrics
Citation Impact
13.2 - 2-year Impact Factor
16.5 - 5-year Impact Factor
3.1 - Source Normalized Impact per Paper (SNIP)
12.7 - SCImago Journal Rank (SJR)
(来源:科学网)
特别声明:本文转载仅仅是出于传播信息的需要,并不意味着代表本网站观点或证实其内容的真实性;如其他媒体、网站或个人从本网站转载使用,须保留本网站注明的“来源”,并自负版权等法律责任;作者如果不希望被转载或者联系转载稿费等事宜,请与我们接洽。