|
|
|
|
|
Direct-seq:单细胞水平分析CRISPR基因编辑筛选实验 | Genome Biology |
|
|
论文标题:Direct-seq: programmed gRNA scaffold for streamlined scRNA-seq in CRISPR screen
期刊:Genome Biology
作者:Qingkai Song, Ke Ni et.al
发表时间:2020/06/08
数字识别码:10.1186/s13059-020-02044-w
微信链接:点击此处阅读微信文章
基于CRISPR基因编辑的正向遗传筛选技术已经广泛应用于“表型(phenotype)-基因型(genotype)”的鉴定研究,常见的研究类型包括鉴定与细胞增殖相关essential gene的负选择实验、与细胞获得抗药抗杀伤能力相关的正选择实验等。其基本原理是,在遗传筛选的起点和终点,细胞群体中 gRNA的种类和频率会发生富集或丢失。由于gRNA本身指代了细胞的基因型,所以研究人员可以据此来推测表型与基因型的对应关系。
但是,这种对应关系是存在局限性的。第一,根据分子生物学的中心法则,遗传信息首先要从DNA传递到RNA,再翻译为蛋白质,并进一步形成独特的细胞表型。然而在目前的研究策略中,基因的RNA表达谱(gene expression profile)这一重要中间层面的信息是完全缺失的。第二,细胞异质性是近年来生物学研究的重点与热点问题。在对筛选终点的细胞群进行分析时,尽管它们呈现出类似的表型,然而它们在分子水平上的异质性是不明确的。
西湖大学近期在开放获取期刊Genome Biology上发表了一项研究成果Direct-seq,该研究开发了一种将CRISPR遗传筛选与单细胞RNA-seq结合的新技术,通过改造gRNA序列,在单细胞水平将细胞的“基因型-基因表达谱-表型”关联起来,从而克服了上述两点局限性。该研究由该校生命科学学院马丽佳实验室完成,该校即将入学的2020级博士研究生宋庆凯、博士后倪科、刘敏博士为并列第一作者。

图1
与之前已经报道的策略不同,Direct-seq在gRNA scaffold序列中加入了一段可以直接被poly(dT)反转录引物高效结合的捕获序列,使得gRNA在细胞中产生的转录本既可以被Cas9结合,实现基因组编辑功能;同时又可以像其他内源表达的mRNA一样,在RNA-seq或单细胞RNA-seq实验中作为转录本的一部分被鉴定。由于只需要对gRNA序列进行简单改造,Direct-seq对于用户使用非常友好。更重要的是,改造后的gRNA scaffold既不影响其本身基因编辑的效率,又可以兼容各类使用poly(dT)作为反转录引物的单细胞RNA-seq建库流程以及商业化平台,包括10x Genomics,Fluidigm C1,DNBeLab C4等。
Direct-seq所采用的捕获序列是一段由A、G两种碱基组成的混合序列(简称“8A8G”),模拟了内源基因转录本的poly(A)尾巴。这种混合捕获序列既有与反转录引物的结合能力,又避免了与Poly(A) Binding Protein结合带来的潜在负面影响。根据实验需要,捕获序列可以添加在gRNA scaffold的Tetraloop, Loop2和Tail三个位置。因此,即使Tetraloop和Loop2被其他aptamer占用(如SAM CRISPR激活系统等),也不影响捕获序列的使用。
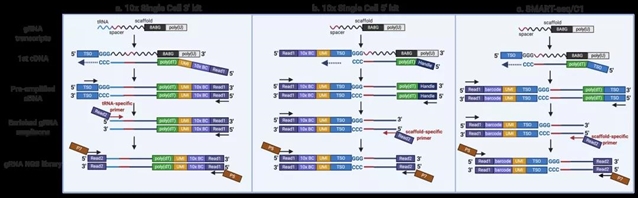
图2
文章同时提供了Direct-seq兼容10x RNA-seq 3’kit,10x RNA-seq 5’ kit和Fluidigm C1/SMART-seq的参考实验流程。据文章报道,在一个使用了10x RNA-seq 3’ v3试剂盒的测试中,由于作者在文库构建过程中使用了巢式PCR特异性富集了gRNA转录本,Direct-seq可以在99.4%的细胞中鉴定出gRNA转录本,在捕获效率上甚至好于10x试剂盒本身提供的基于Featured Barcode的技术。
招募令
西湖大学马丽佳实验室关注基因编辑和组学技术开发与临床应用。目前有博士后名额开放申请。
欢迎免疫学、胚胎学、分子生物学、生物信息学和微流控技术等研究方向的申请人咨询和加盟,请直接邮件联系[email protected]

图3
Genome Biology covers all areas of biology and biomedicine studied from a genomic and post-genomic perspective. Content includes research, new methods and software tools, and reviews, opinions and commentaries. Areas covered include, but are not limited to: sequence analysis; bioinformatics; insights into molecular, cellular and organismal biology; functional genomics; epigenomics; population genomics; proteomics; comparative biology and evolution; systems and network biology; genome editing and engineering; genomics of disease; and clinical genomics. All content is open access immediately on publication.
摘要:CRISPR-based genome perturbation provides a new avenue to conveniently change DNA sequences, transcription, and epigenetic modifications in genetic screens. However, it remains challenging to assay the complex molecular readouts after perturbation at high resolution and at scale. By introducing an A/G mixed capture sequence into the gRNA scaffold, we demonstrate that gRNA transcripts could be directly reverse transcribed by poly (dT) primer together with the endogenous mRNA, followed by high-content molecular phenotyping in scRNA-seq (Direct-seq). With this method, the CRISPR perturbation and its transcriptional readouts can be profiled together in a streamlined workflow.
(来源:科学网)
特别声明:本文转载仅仅是出于传播信息的需要,并不意味着代表本网站观点或证实其内容的真实性;如其他媒体、网站或个人从本网站转载使用,须保留本网站注明的“来源”,并自负版权等法律责任;作者如果不希望被转载或者联系转载稿费等事宜,请与我们接洽。