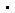ASGR1调控脂质稳态示意图
中科院广州生物医药与健康研究院研究员李尹雄团队发现血脂稳态调控新机制,一种肝细胞膜上的特有蛋白质,去唾液酸糖蛋白受体1(ASGR1), 通过胰岛素诱导基因1的编码蛋白(INSIG1),量化调控固醇类反应元件的结合蛋白(SREBPs)的胞内运输和活化,协调低密度脂蛋白受体(LDLR)在肝细胞膜上的含量以及肝细胞内吞清理低密度脂蛋白(LDL)的速率,动态维持甘油三酯和胆固醇在血液中的水平,为降血脂的药物研发提供了新靶点。相关研究10月9日在线发表于《临床研究杂志》子刊《机理解析》。李尹雄为论文的通讯作者,徐莹莹、陶家旺和于小瑞为论文共同第一作者。
高血脂高血糖相关的糖尿病和心脑血管病严重威胁全球人群的生命和健康,究其根源是糖脂代谢出现了紊乱,早期表现为脂肪肝、高血糖和高血脂。2020年发表的《中国心血管健康与疾病报告》显示,我国成人代谢综合征的患病率为24.2%、糖尿病患病率为11.2%、心血管病患病率为23%(3.3亿);中国心血管病死亡占城乡居民总死亡原因的首位,农村为46.7%、城市为43.8%。自2004年以来,急性心肌梗死、缺血性脑卒中和颅内出血住院费用的年均增长速度分别为26. 9%、18.7%和14%,远超我国GDP的增长速度,代谢病和心脑血管病给个人、家庭和社会带来了巨大的经济和精神负担,该状况在未来数年将会变得更加严峻。
目前,国际上普遍使用的降血脂药物是它汀类药物,该类药物的靶点是合成胆固醇的关键限速酶(HMG-CoA还原酶),它汀类药物的使用确实能降低冠心病的发病率和死亡率,但仍有其局限性,一方面存在对它汀类药物不敏感的患者;另一方面,长期使用他汀类药物带来的副作用,包括高血糖、横纹肌溶解、肝功能损害和记忆和认知障碍。因此,急需寻找发现新的降血脂策略和新的药物靶点。
2016年底《新英格兰医学杂志》报道了一项基于百十余万份的多国人群全基因组测序和心血管病关联性的调查报告,发现多个基因与心血管发病趋势和血脂水平密切相关,其中一个叫Asgr1的基因令人惊讶,该基因在健康人群中存在一种极其罕见的12个碱基缺失的杂合突变,该突变的携带者其血浆非高密度脂蛋白胆固醇水平比正常人群低9%,甘油三酯低6.1%及冠心病发病风险降低34%。于是从基础研究到药物研发,从科研单位到制药公司,都高度关注ASGR这个降脂的潜在靶点,同时一场全球性竞赛在静悄悄地拉开了序幕,各路人马试图确证该靶点是一个全新可靠的降脂药物靶点,寄以厚望的工作聚焦在能否利用小鼠模型重复人群中的表型,并阐明其调控血脂稳态的深层分子机理。
肝脏是血脂及载脂蛋白代谢的中心,ASGR1是评判肝细胞成熟度的指标之一,成熟的肝细胞特异表达ASGR1并定位于肝细胞膜上,负责去唾液酸化糖蛋白的内吞和清除, 已有发现表明,ASGR1介导衰老血小板和一些去糖基化血浆蛋白的清除和降解,并且该蛋白参与了自身免疫疾病、肿瘤转移和肝特异性病毒的入侵等病理过程。
为了确证ASGR1调控脂代谢并阐明其机理,李尹雄团队利用基因编辑技术分别建立了 ASGR1 基因敲除的小鼠和细胞模型。实验结果显示Asgr1敲除小鼠重复了人群携带者的表型,各项血脂均有不同程度降低,其中低密度脂蛋白胆固醇的降低最为显著,敲除一半和全部敲除ASGR1导致的表型程度呈现剂量效应,确证ASGR1是一个理想的药物干预靶点。
Asgr1敲除小鼠血脂的降低是由于肝细胞对低密度脂蛋白的吞噬增多和分泌减少两方面因素造成的。ASGR1蛋白缺失后,一方面,肝细胞内调控LDLR降解的关键蛋白PCSK9在转录和翻译水平均降低,进而导致LDLR蛋白水平增多,促进肝细胞对LDL的吞噬;另一方面,ASGR1蛋白缺失后,肝细胞内组装VLDL/LDL的关键蛋白MTTP显著降低,导致LDL的分泌减少。
进一步机理刨析发现ASGR1的降低,导致INSIG1蛋白增高,INSIG1将调控脂代谢的关键转录因子SREBPs锚定在内质网上,减少了SREBPs蛋白的高尔基运输、剪切、成熟、入核以及其把基因的表达,这些基因的功能涉及脂质合成、转运和代谢,其中的关键靶基因PCSK9和MTTP转录水平降低,导致了肝细胞吞噬LDL增多同时分泌LDL减少。分别在Asgr1-/—小鼠和ASGR1-/—细胞上敲降INSIG1和回补表达ASGR1,均能有效逆转ASGR1敲除引起的脂质吸收增多以及分泌减少的表型。揭示ASGR1通过INSIG1调控SREBPs的活化水平是维持血脂稳态的关键调控环节,ASGR1-INSIG1-SREBPs调控轴的发现为降血脂药物的研发的提供了理论基础和新的靶点。(来源:中国科学报 朱汉斌 黄博纯)
版权声明:凡本网注明“来源:中国科学报、科学网、科学新闻杂志”的所有作品,网站转载,请在正文上方注明来源和作者,且不得对内容作实质性改动;微信公众号、头条号等新媒体平台,转载请联系授权。邮箱:
[email protected]。