|
|
|
|
|
科学家制备铁催化噻吩C–H/C–H区域选择性偶联的新型有机半导体材料 |
|
|
具有区域选择性的噻吩-噻吩偶联可以说是有机电子材料中最重要的转化之一。但目前传统的偶联方法由于涉及到氧化剂的使用常常限制了合成效率和对氧化剂敏感的底物的适用性。
日前,日本东京大学的尚睿/中村栄一团队发展了一种基于铁催化噻吩C–H/C–H区域选择性偶联的新型小分子和聚合物有机半导体材料制备技术。相关研究成果以“Iron-catalysed regioselective thienyl C–H/C–H coupling”为题,于2021年7月19日发表在Nature Catalysis上。
出于步骤经济性和原子经济性的考虑,由过渡金属催化的直接 C–H 键活化反应在共轭材料科学中起着越来越重要的作用。在几种类型的直接 C–H 偶联方法中,如果可以实现高区域选择性和高效率,C–H / C–H 偶联是连接两个π单元的最直接方式。而π分子单元的直接 C–H / C–H 缩聚则可以被称为高分子电子材料的合成方法中的“圣杯”。传统的通过阳离子或自由基阳离子机制进行C–H/C–H 偶联的方法(例如 Scholl 反应和氧化芳香偶联)具有非常有限的底物范围和较差的区域选择性,这也限制了它们在能源器件和电子材料的合成中的应用。
虽然过渡金属催化的C–H/C–H 偶联在过去十年中取得了重大进展,但具有完全区域选择性控制并且能够兼容电子材料中必要分子结构的催化方法仍然未被开发。此外,若能够使电子材料中的π结构进行C–H 缩聚,这对催化剂的效率也是一个相当大的挑战。对于过渡金属催化的电子材料共轭π单元的直接C–H/C–H偶联,主要挑战来源于富d电子的后过渡金属与共轭单元的强金属-π相互作用,因为这会干扰到 C–H键与金属中心的相互作用。与苯基序不同的是,用于电子材料的大共轭π-基序是电子的供体和受体并且对氧化还原特别敏感,进而干扰到需要用氧化剂的氧化催化循环。这些氧化剂可以直接从π单元中夺取电子,尤其是当有供体π结构的分子参与的情况下。
根据研究团队过去探索的铁作为 C–H 键活化催化剂的经验,底物和配体与铁之间的电荷离域增强了反应性。于是研究人员设想用于C–H活化的Fe(III)/Fe(I) 催化循环可以解决这些问题,从而允许在共轭π结构上进行有效的C–H/C–H偶联。此方法具有以下三个优点:1) d5-Fe(III) 与π结构的相互作用较弱,因此 C–H 键的裂解通过去质子的方式进行。与富含 d 电子的晚期过渡金属(例如 d8-Pd(II))相比,这对各种π单元的干扰较小。2) 非常弱的氧化剂(比如 1,2-二氯烷烃)就足以将 Fe(I) 转化为 Fe(III)(例如Fe(III)/Fe(I)的氧化还原电位与标准氢电极相比小于0.55 V 而Pd(II)/Pd(0)的则为0.92V)。3) d5-Fe(III) 很容易从产品中去除,而不会对材料性能产生不利影响。
因此这里该团队开展了一种具有区域选择性的铁催化噻吩C–H/C–H 偶联的方法(图1b, I 和II)。草酸二乙酯(DEO)可作为一种潜在的氧化剂与Al(III)结合,通过接受两个电子来再生三价铁催化剂(图1b, III)。这可能是一种环内的电子转移进而形成一种热力学上稳定的烯二酸铝五元环(图1b, IV)。研究人员还设计了一种廉价易得的三齿膦配体TP1以控制铁中心的配位环境(图1c)。
图1:a,氧化条件下的噻吩C-H/C-H偶联。b,铁催化的区域选择性噻吩 C-H/C-H 偶联,通过用 AlMe3 进行去质子 C-H 活化和用 DEO/Al(III) 再生 Fe(III) 催化剂。c,最有效的TP的结构及其合成所需的底物。
图2总结了关键的反应参数。经过研究人员筛选发现,廉价易得的草酸二乙酯是最有效的电子受体。此反应的一个典型例子如图2a所示,以苯并噻吩1作为底物,使用催化量的Fe(acac)3和共轭三齿膦配体TP2(该配体此前曾被该团队用于铁催化的C-H甲基化反应),当量的AlMe3以及50 mol%的DEO,70摄氏度下在四氢呋喃和甲苯的混合溶剂中反应,得到了C2位点反应的二聚物。而C3位点活化的产物和甲基化的产物都没有形成。
随后研究人员使用了二苯基乙二酮来替代DEO,得到了产物2和苯偶姻(图2b)。这证明了反应过程中烯二酸酯IV的形成,为DEO作为二电子受体提供了证据。研究人员也曾尝试检测IV和水解后的乙醛酸乙酯,但均未成功,这可能是由于铝物种的高聚集性和高亲氧性导致的,而这种聚集性也意外地为产品的分离提供了好处。
图2:a,标准反应条件下苯并[b]噻吩的铁催化区域选择性噻吩C-H/C-H偶联。b,二苯基乙二酮的二电子还原产生的苯偶姻的检测。c,关键反应参数表。
图2中的数据很好地说明了此反应的一些特征。首先9,10-二氢蒽的加入并未对产率造成任何影响,这排除了氧化性自由基或自由基正离子参与的可能性(entry 2)。相比于其他氧化剂,DEO被证明为效率最高的(entry 6-10)。AlMe3的参与对反应也是必要的(entry12),而使用AlEt3或MeLi,MeMgBr,MeZnBr时,则并没有产物生成。推测AlEt3参与时由于乙基β氢消除而产生的铁氢络合物抑制了反应的进行。而后三者试剂则是因为它们的强亲核性与草酸盐不相容,以及它们的强还原性会削弱三价铁的活性。TP2的参与对反应也是必要的(entry 15),当使用PPh3,dppbz和TetraP作为配体时反应则也不会进行(entry16-18)。但TP1和TP2在二聚反应是能力是相当的(entry19),但对于缩聚反应而言TP1反而是必要的(图3)。根据以往的报道,三齿膦配体和铁的络合物会留出三个位点参与催化,因此TP的大体积所带来的位阻效应是不容忽视的,这点从entry10和entry11的实验也得到了证实:1-十八烯的加入抑制了反应,但反式1,2-二苯乙烯的加入对反应影响甚微。
随后研究人员发展了通过C–H活化的噻吩的缩聚反应。对于缺电的聚合物的合成,以往只有极个别的报道使用了金属催化的碳氢活化反应,而对于供电型的半导体聚合物而言,以往并没有类似的报道。这里研究人员选择了2,7-二噻吩咔唑3作为反应物,成功实现了一种噻吩-咔唑聚合物4的合成,此类结构常用作为有机薄膜晶体管的材料(图3a)。随后研究人员探究了各类三齿配体对反应的影响(图3b),其中TP的电性对起着关键作用。若在中心磷原子上引入一个富电的芳基,则会明显降低产物的聚合度(entry1-5)。而对于TP7而言,富电的吲哚基团和N-甲基的取代基所带来的位阻效应是降低反应性的主要原因。而TP1和TP8在反应中都有着优异的表现。动力学实则验证实了反应是一个逐步聚合的过程(图3c)。
图3:a, 利用铁催化的具有区域选择性的噻吩C–H/C–H缩聚反应获得DTC聚合物。b. 配体对缩聚反应的影响。c, 化合物5的Mn和Mw/Mn值作为单体转化率的函数。
最后该团队进行了偶联反应和缩聚反应的底物拓展。
图4说明了噻吩C–H/C–H偶联的底物范围。该反应对噻吩环以及苯并[b]噻吩(2)、噻吩(6和7)和苯并 [1,2-b:4,5-b]二噻吩(11)的二聚体展现出独特的 C2(或 C5)选择性,并以极好的收率获得了产物,并且几乎没有发生C2甲基化(仅对18-20检测到了产率<1%的甲基化产物)。C3位上的取代基会抑制相邻 C2 位置上的 C-H 偶联,比如对于C3位被己基取代的底物而言,二烷基化四噻吩(9)是唯一的产物。而由于 3,4-取代基的位阻,富含电子的 3,4-亚乙基二氧噻吩(18)则以中等产率得到了偶联产物。
对于光电材料中常见的π基序,如苯并[b]呋喃(10)、亚乙烯基(12)、芴(16)、咔唑(19和20)和富电子三芳基胺(21和22),也以很好的产率得到了相应的产物。对于12而言,反应以高收率获得了低聚噻吩亚乙烯基产物,并且未观察到E-亚乙烯基结构的立体异构化。该反应同样可以耐受芳烃上的三异丙基甲硅烷基(11-13)、三丁基锡(14)和频卡硼酸酯(15)。而对于在传统氧化芳烃偶联条件下易关环的17而言,在此条件下其多环芳烃序列仍然保持完整。用于钙钛矿太阳能电池的二噻吩22也以极高的产率被得到。此外反应可以很容易放大到克级,如化合物19。
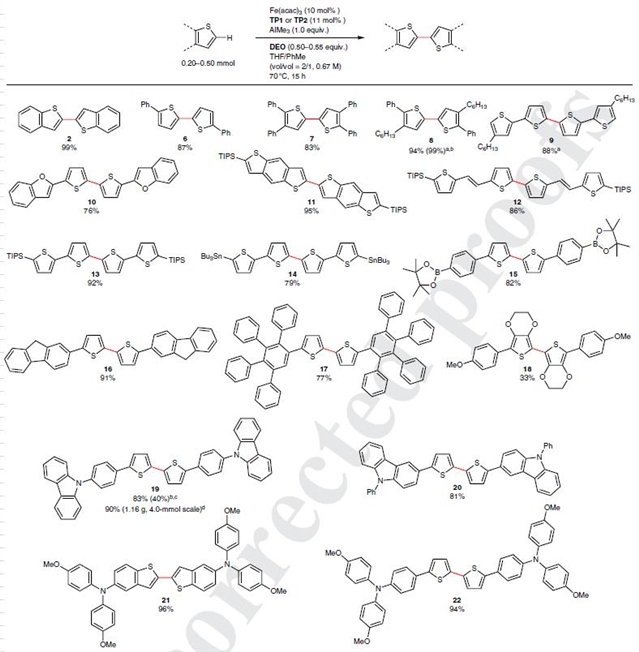
图4 噻吩C–H/C–H偶联的底物拓展

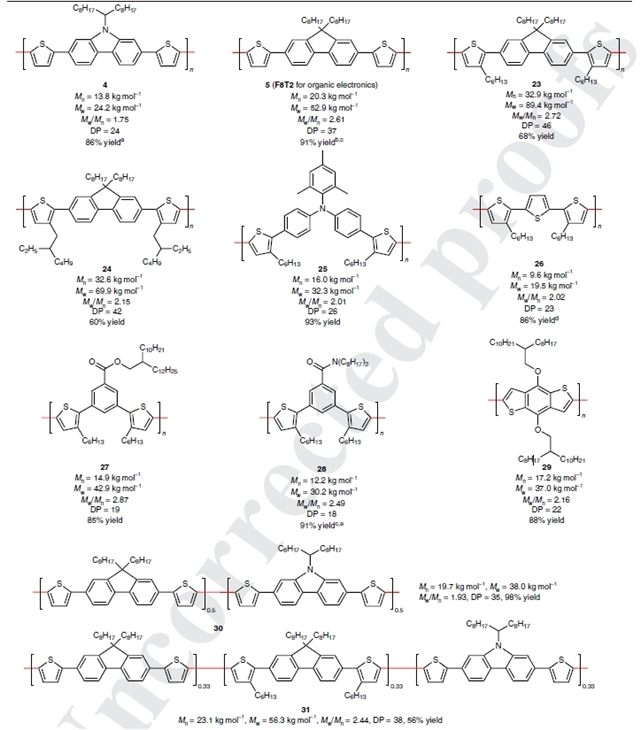
图5:铁催化区域选择性噻吩 C–H/C–H 缩聚的底物拓展
图5展示了缩聚反应的底物适用性,而这些产物在传统氧化芳香C–H/C–H偶联或钯催化的C–H/C–H 偶联的条件下是无法得到的,且出色的 C2 区域选择性避免了C3位支化聚合物的污染。
聚(9,9-二辛基芴-alt-联噻吩)(5;缩写为 F8T2)是一种商品化的半导体聚合物(数均分子量Mn = 16 kDa;多分散指数PDI = 2.76),且常可能含有200 ppm 的Pd,它被广泛用于有机发光二极管、光伏和场效应晶体管中。这里以 91% 的分离产率得到了5,且具有大的分子量(Mn = 20.3 kDa;聚合度= 37)和 2.61 的PDI(该值表明与市售 F8T2 相比分子量分布更窄)。通过用二氧化硅金属清除剂的处理,F8T2 中的铁含量可以降低到43 ppm。研究人员还通过在单体上引入烷基取代基来增加 F8T2 的溶解度,将Mn值增加到了33 kDa,聚合度增加到了42-46(23和24)。
研究人员还以93%的产率制备了一种含有 3-己基噻吩和三芳基胺基序(25)的新聚合物,聚合度为 26,PDI为2.01。25的电离电位(–5.4 eV;薄膜)接近甲基铵碘化铅钙钛矿的电离电位,表明其在太阳能电池中具有潜在的应用。
利用本方法优异的的区域选择性,还可以从 3,3-二己基-2,2:5,2-三联噻吩单体合成聚噻吩(26)。该聚合物(26)平均含有69个噻吩单元,且在特定位点带有己基侧链。包含酯和酰胺基团作为电子受体的π基序的聚合物也可以以高产率获得(27和28)。以4,8-二烷氧基苯并[1,2-b:4,5-b]二噻吩为单体,还可以得到聚合度为22且PDI为2.16的聚合物29。
最后通过两种和三种不同单体的共缩聚得到了相应的聚合物(30和31),且聚合物中单体的比例与初始反应物比例相同。目前该聚合方法的限制是不适用于交替共聚。研究人员称,通过使用他们报道的在关键基团存在下的瞬态连接策略,进一步开发两种不同底物之间无自偶联的C–H 活化/交叉偶联反应,有望实现交替的C–H/C–H 共聚反应。
综上,尚睿/中村栄一团队使用两种地球上最丰富的两种金属(即铁和铝)来合成π共轭小分子以及在有机电子器件中具有重要应用的大分子。三齿膦配体TP和DEO/Al(III) 的组合对于控制有机铁中间体的价态和电子自旋态起着重要作用。这里发展的C–H/C–H 缩聚反应对相应聚合物的合成方法起了补充作用,该团队还将据此进一步开发相关的高效铁催化剂。(来源:科学网)
相关论文信息:https://doi.org/10.1038/s41929-021-00653-7
特别声明:本文转载仅仅是出于传播信息的需要,并不意味着代表本网站观点或证实其内容的真实性;如其他媒体、网站或个人从本网站转载使用,须保留本网站注明的“来源”,并自负版权等法律责任;作者如果不希望被转载或者联系转载稿费等事宜,请与我们接洽。